GenIO assists medical doctors in the clinical genomics diagnostic process. GenIO prioritizes the most probable variants causing a rare genetic disease using the genomic and clinical information provided by a medical practitioner.
GenIO stands for Genomic Input Output, and it's a clinical genomics web server designed to help geneticist diagnose rare diseases based on the patient’s genetic makeup and clinical information. GenIO annotates, classifies and filters variants identified in whole genome, whole exome or target sequencing studies, and identifies candidate genes associated with the patient’s symptoms.
GenIO key features:
Simple, intuitive and user-friendly interface for medical doctors
Easy gene filtering by symptoms, suspected disease, and complementary findings with controlled vocabulary
Further filtering options by gene list and frequency scales
Identification of rare disease variants in inheritance models
Variant clinical classification following ClinVar, M-CAP, and ACMG/AMP standards and guidelines
Secondary findings identification
VCF file full annotation
GenIO was made to systematize the crucial and time consuming task of clinical genetics data annotation and filtering. Most geneticists are doing this task manually with bioinformatics support since the first clinical use of whole-genome sequencing, published in "Human genetics: Genomes on prescription" Nature, Vol. 478, No. 7367. October 2011. This work has inspired the creation of GenIO.

The user inputs the patient's genetic makeup as a VCF file along with the symptoms observed, a suspected disease and complementary findings from the patient´s clinical records.
All this information from the patient's phenotype is fed to the Phenolyzer, which gives a list of candidate genes whose alteration is associated/linked to the symptoms/diseases given. In addition, the user can provide their own list of suspected genes.
The candidate inheritance model variants are identified by running the VCF file through Annovar's table_reduction script for both a recessive and dominant model, and then matching the genes with variants to the genes listed by the Phenolyzer.
A deeper analysis is made to identify variants with potential functional pathogenic effects that may be related to disease by filtering variants in genes involved in Mendelian disorders (in the OMIM database), with impact on the gene product (nonsense and frameshift mutations, splice site alterations, loss of stop codons, non-synonymous substitutions and codon insertions and deletions), with a gnomAD exome allele frequency < 1%, and with a clinical significance of pathogenic or likely-pathogenic obtained either from the ClinVar database or by the Mendelian Clinically Applicable Pathogenicity (M-CAP) classifier.
Finally, GenIO creates a secondary findings minimum list that includes deleterious variants found in 59 medically actionable genes (ACMG SF v2.0) recommended for return in clinical genomic sequencing studies.
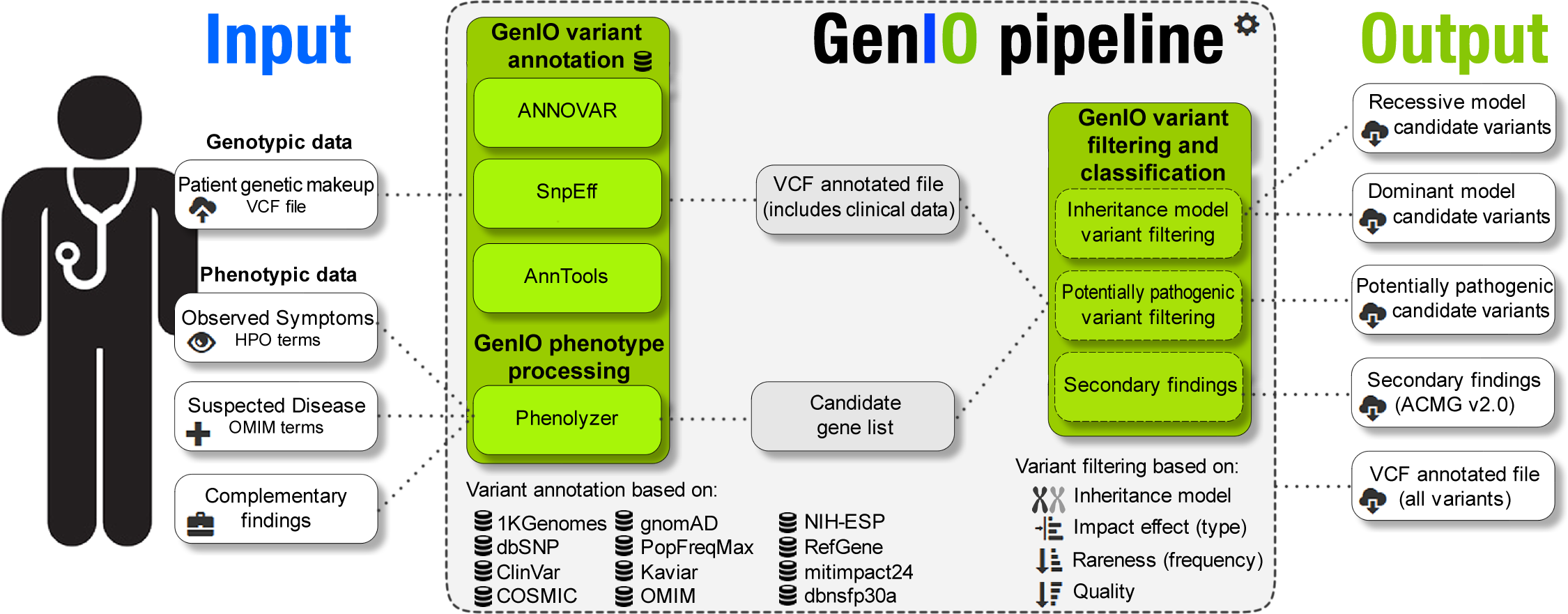
Third-party programs used:
Annovar (v.2017Jul17)
AnnTools (v.1.1)
SnpEff (v.4.2)
Phenolyzer (v.1.0.5)
The VCF input file must comply with the standardized VCF format version 4.0 or higher (http://www.internationalgenome.org/wiki/Analysis/variant-call-format), must be based on the GRCh37/hg19 human reference genome assembly, and must have some fields completed (with the exception for the INFO and ID fields that can be empty), since all the information is required at the different filtering processes in our pipeline. The VCF header should contain the format information, and the column names and order must be: CHROM, POS, ID, REF, ALT, QUAL, FILTER, INFO, FORMAT, SampleID. These columns must be tab separated, have each the proper data type, and have no duplicated variant lines (that is same chromosome, position, reference and alternate info). Please note that only variants that have passed all the quality controls, and hence have a PASS value in the FILTER field, will be taken in consideration for the analysis.
GenIO's input interface has an auto-complete function for Human Phenotype Ontology (HPO) and Online Mendelian Inheritance in Man (OMIM) terms. The user just needs to start typing the symptoms, disease and/or complementary findings to select the correct term.
Although this is not a mandatory input, this is crucial information for the identification of the variants most likely associated with disease. GenIO will annotate and clinically classify the variants in the genotype VCF file anyway. Providing the disease/phenotype terms will certainly improve GenIO's performance.
The input will be accepted, but it will be of no use for GenIO. (Be careful with misspellings, it's better to start typing and use the auto-complete function to cast away any doubts)
There are different databases integrated into the pipeline, each providing different kind of information associated to the variants, being of computational, biological or medical nature. This is the list of the most important ones:
RefGene (v.20151211)
NHLBI-ESP (v.2)
1KGenomes (v.2015Aug)
dbSNP (v.avsnp150)
ClinVar (v.20170905)
COSMIC (v.70)
gnomAD (20170311)
OMIM (v.2.1.2-2016-06-11)
M-CAP (v.1.0-2016-10-24)
Reference Genome (GRCh37/hg19)
When the analysis is finished the user receives an email notification with a link to the results page, which includes:
List of rare variants obtained by applying the recessive inheritance model filter
List of rare variants obtained by applying the dominant/de novo inheritance model filter
Enriched full annotated VCF file with up-to-date clinical genetic information
List of potential pathogenic variants obtained by applying filters, ACMG/AMP, M-CAP and ClinVar classification
List of rare variants found in the entered list of genes of your interest
Secondary findings obtained by following the ACMG recommendations
Along with the possibility of downloading all these files in a single zipped file.
The job will be avariable in our servers for a period of one month.
Link to the ACMG/AMP standards and guidelines for the interpretation of sequence variants
Link to the ACMG recommendations for reporting of secondary findings
If you find GenIO useful, please consider citing our publication:
GenIO: a phenotype-genotype analysis web server for clinical genomics of rare diseases.
Daniel Koile, Marta Cordoba, Maximiliano de Sousa Serro, Marcelo Andres Kauffman and Patricio Yankilevich
BMC Bioinformatics 2018, 19:25 | Published on: 27 January 2018.
